


Circling around patient care and improving overall healthcare services, the European Parliament has set new requirements for medical device and in vitro diagnostic manufacturers that distribute products in the EU. However, medical device manufacturers have realized that they are bound to face many challenges in order to make their products market-ready, not to forget the gigantic task of implementing new protocols in a timely manner, which will not be easy.
Need for a comprehensive updated medical device regulatory system
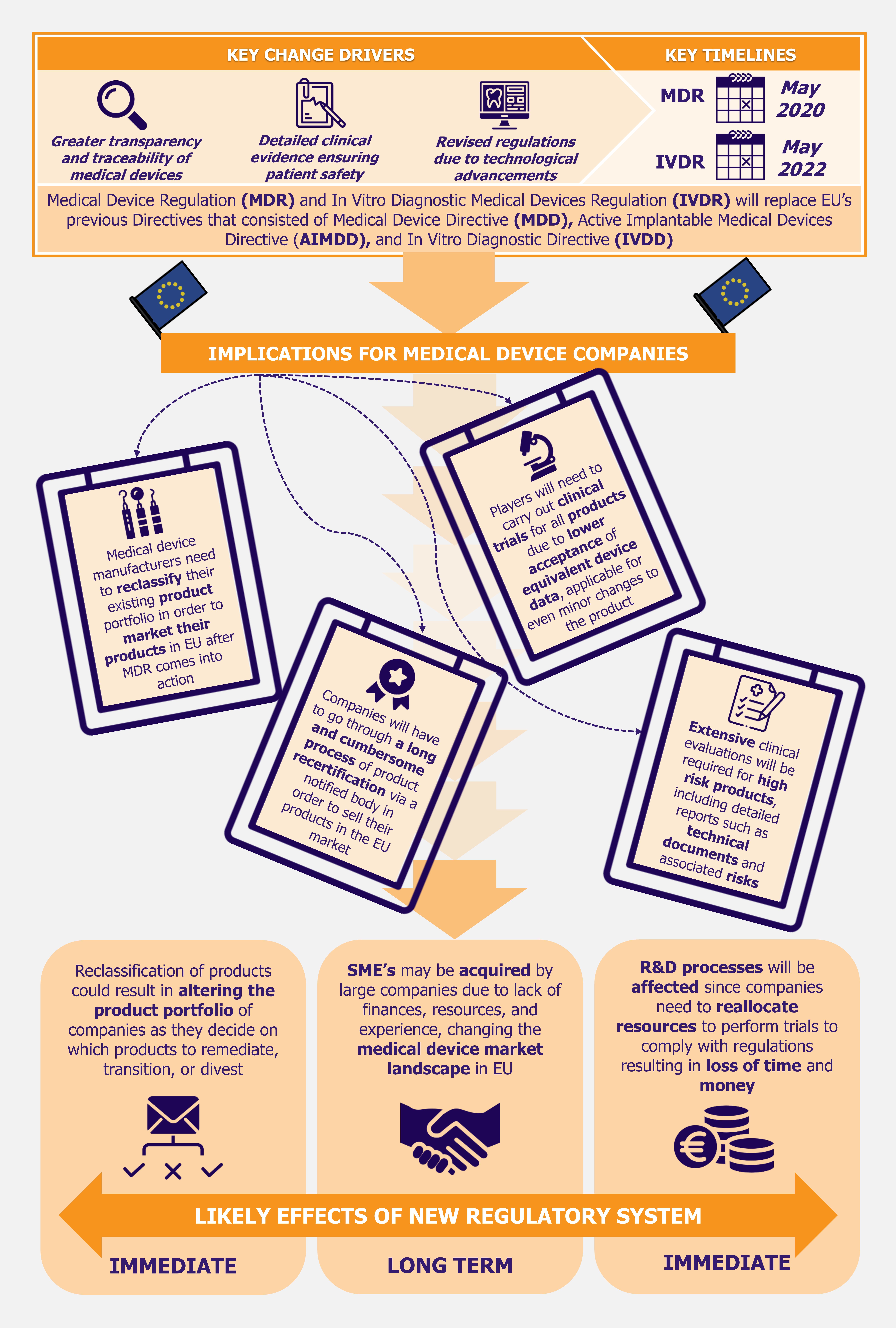
EU’s Medical Device Regulation (MDR) and In Vitro Diagnostic Medical Devices Regulation (IVDR) were made official in May 2017, with transition period of three years (fully applicable from May 26, 2020) for the former and five years (fully applicable from May 26, 2022) for the latter. These regulations will replace EU’s previous directives: Medical Device Directive (MDD), Active Implantable Medical Devices Directive (AIMDD), and In Vitro Diagnostic Directive (IVDD).
The need for new regulations of medical devices in EU arose from the growing demand for technologically advanced medical products which necessitated more stringent monitoring of these devices to ensure a high level of efficacy and safety among patients.
Unlike earlier version of the regulations where the main focus revolved around the pre-approval stage of medical device manufacturing, the new regulatory guidelines promote an overall product-life cycle approach, focusing on both device safety and performance.
Enhanced supervision, easy documentation of devices, more stringent clinical evidence requirement, and increased supervision on part of authorities providing medical device certifications are some of the key changes in MDR as compared to the EU’s previous directives.
Bumpy road ahead for medical device manufacturers
Reclassifying existing product line-up
Based on the risk factor, changes have been made to the way medical devices are classified. Under MDR, the number of classification rules has expanded from 18 to 22 intensifying the task of product re-classifications by the manufacturer.
For instance, products using software for monitoring purposes being implanted in the body has been reclassified to higher-risk class (from Class I to Class III) which would now require conformity assessment by a notified body (NB – an organization that assess the conformity of medical devices before they are placed on the market), unlike earlier, when Class I products did not require assessment via a NB. This is going to burden players with increased operational costs; thus, it is imperative that the manufacturers familiarize themselves with the classification changes and study the impact on their product portfolio.
New products are also being added to the list of medical devices that earlier were not part of the medical device regulatory framework. For instance, products manufactured utilizing human tissues or cells and devices incorporating nanomaterial, under new regulations, will be considered medical devices. Manufacturers of such products have work cut out for them – from conducting clinical investigations, preparing technical documentation and evaluation processes, to product certification. Though such products could only form a very small percentage of the company’s product range, the task to make them available in the market is great, especially under current circumstances.
Manufacturers who do not comply with the new regulations will no longer be able to market their products in Europe. Thus, a robust strategy in terms of resource allocation, time management, and budget is an absolute must for manufacturers to make this transition possible.
Distress over notified bodies
Strict parameters are also being applied on NBs. Since all devices will require new certification from a NB, only designated NBs will be able to certify a device. The designation process is a complex procedure as it involves audits and corrective actions (once a NB expresses interest). However, while the medical device manufacturers have been in the process of switching to newer protocols since mid-2017, the first designated NB (BSI United Kingdom, the national standards body of the UK) was announced in January, 2019, almost 18 months after the regulations were announced and 14 months into the formally started designation process.
Such time-consuming process raises concern among medical device companies about the ability to complete the necessary conformity assessments within the allotted time. The huge task of recertifying medical devices with only a handful of designated NBs is a cause of worry for companies, as it could potentially result in significant backlogs as the last date approaches. However, there is only so much companies can do – even though they are proactive to comply with the new regulations much ahead of the deadline, poor process planning and lack of supporting bodies (notified bodies in this case) results in a long halt for these players.
The companies are heavily dependent on NBs for auditing and product certification, and the insufficient number of designated bodies adds to the risk of many devices being non-compliant according to new regulations. As of May 2019, less than 40 NBs have filed application for designation procedure (out of 58 designated NBs under the directives); only two have actually received a designated status – BSI UK and Germany based TÜV SÜD Product Service GmbH Zertifizierstellen (certification received in May 2019). With very little time at hand to reassess and rectify issues (if any), this could jeopardize the product availability in the market, resulting in not only risking the patients’ life (due to non-availability) but also in huge financial losses for the players.
Detailed clinical evaluation of medical devices
Owing to reclassification of product categories, many devices will require changes to their existing clinical evaluation reports, another challenge for medical device manufacturers. Manufacturers that have not previously been required to perform clinical testing will have to do so now. For instance, mechanical heart valve sizers will be moved up from Class I to Class III, and unlike in MDD where clinical evaluation was based on literature analysis, new evaluation of valve sizers will require clinical investigation. This will require a huge deal of additional time, money, and expertise, further burdening the device manufacturers.
Medical devices already in the market that remain untouched by the reclassification criteria will still require reassessment of clinical data. If the data do not meet the new requirements, devices will need to undergo additional testing to be recertified, increasing the expense for manufacturers.
MDR also calls for inclusion of risk management within the clinical evaluation expecting clinical risks to be addressed in clinical investigations and evaluation studies – adding another task to the long list of activities to be accomplished before MDR fully rolls out.
Explore our other Perspectives on medical devices markets
Comprehensive demonstration of equivalence data
Unlike MDD, where device manufacturers were able to use clinical data of an equivalent device for their own product registration, under MDR, equivalence is going to be less accepted, particularly for higher risk devices.
There are two ways out – manufacturers can either conduct their own trials not having to deal with the equivalence commotion or they can demonstrate that they have access to the equivalent device (with respect to technical and clinical properties) data. The latter is highly unlikely to happen considering equivalent device would typically belong to a competitor unwilling to grant such access. Thus, with stern requirements for comparative evaluations, more effort, planning, money, and resources will be needed for device manufacturers to demonstrate product safety and performance.
As new medical devices are developed, multiple small incremental improvements (minor changes in design, addition or subtraction of small hardware parts such as bolts or screws) happen over time. Once the device is already in the market, it is practically impossible to conduct a re-trial to gain approval for such small changes. An expected solution to this would be a provision to accept such minor changes through pre-clinical evidence or prior trial results. However, with equivalence testing being reduced drastically under MDR, unless a solution for such cases is offered, manufacturers will have to conduct re-trial and re-document everything, which would result in significantly increased cost. Another issue that could arise from such situations is the reduction in R&D activities inclined towards product improvement.
Trouble galore for SME’s
While making amendments and prioritizing to comply with new regulations seems to be the top most priority for medical and diagnostic device manufacturers, it seems SMEs will be dramatically more impacted than large players – in Europe, a small-sized company employs less than 50 people and has a turnover of less than or equal to €10 million while a medium-sized company employs less than 250 people and has a turnover of less than or equal to €50 million. Owing to the increase in cost, time, and resources associated with the process, the new regulations may put smaller companies under pressure, possibly resulting in altering (such as merging with or being acquired by larger companies) the European medical device market structure, currently dominated by SMEs – there are nearly 27,000 medical technology companies in EU, 95% of which are SMEs.
SMEs also need to be more vigilant when it comes to being associated with a designated NB as not all currently functioning NBs are expected to get a designated status. With their already dwindling numbers married with an increased demand for their services, once the new regulations roll out, it is quite possible that small manufacturers are orphaned since NBs could be partial towards larger players and prioritize them over other small and medium players.
Smaller players will not only have to hire additional personnel for dealing with regulatory issues but also employ clinical trials specialists (for documenting insights to be presented and approved by the NB) for launching products in the market which means higher costs. Adjusting budgets to keep costs under control would hamper other critical business operations, e.g. reduce R&D activities or cut the number of products being launched in the market.
As a step to overcome these issues, players with limited financial resources should strategically study their product portfolio to determine which products are worth investing in for MDR compliance. For doing this, they should lay out a detailed plan for each product and decide whether to remediate, transition, or divest.
It is also advised that SMEs should devise a clear step-by-step approach plan to ensure compliance. As an alternative to hiring transition specialists, they could engage employees from various functions within the organization to take responsibility for specific processes thus keeping costs in check.
EOS Perspective
The changes and revisions required to be carried out under MDR are company-wide and require significant investment to plan and execute. This will lead to players devising a business strategy based on assessing risk associated with product portfolio (whether some products need to be pulled out from the market and what effect it would have on future revenue) or looking for acquisition partners. Based on these decisions, the medical device market topography in EU is expected to see some major changes in the coming years – small companies looking for partners to get acquired or for new partnership with a service provider (specializing in regulations compliance). This will also result in organizational restructuring, revamping design processes, and systems implementation.
Companies have to make crucial decisions around the product portfolio. For some of the already existing products, if reclassified, the cost of compliance could be much higher than actual market returns. In such cases, manufacturers may be compelled to pull away such products from the market resulting in high healthcare costs and ultimately burdening the patients, who (theoretically) form the center point of the MDR. Though this is unlikely to happen at a large scale, since there are always alternative products available, it cannot be denied that this may be a major loophole in MDR requiring immediate attention.
Since SMEs drive the EU medical device market, as an immediate consequence, MDR is not likely to have any positive effect on these players other than distorting their business operations. However, it can only be anticipated that, with time, MDR may adapt and amend to offer some relaxation in provisions especially for small and medium-sized players. Nonetheless, MDR also brings an opportunity for such players to audit their current offerings and come out with an enhanced product portfolio, which could be an opportunity to be capitalized on in the distant future.
Modifications being made in the functioning of NBs are also likely to have an impact on the device manufacturers. For high risk devices, manufacturers may expect deeper scrutiny of design records and data files leading to providing more credentials, in case any query arises. This, along with long wait time for product review (due to reduction in the number of designated NBs) and limited availability of resources (again on account of NBs), could lead to unknown delays for obtaining product re-certification. Thus, companies need to chalk out their market strategies very effectively and be prepared to address any concern that rises during product reviews.
The aim of implementing new regulations is to bring a transparent and robust regulatory framework for medical devices. However, there is no assurance that the new regulations are completely accurate and will apply seamlessly to live case scenarios. Therefore, once implemented, there is a possibility that MDR may see revisions in the initial months of coming into action.
These changes, though certainly positive from a healthcare point of view, are enormous. Transitioning to meet the new standards within the stipulated time frame is challenging for manufacturers. Not adapting to the changes is not a choice for manufacturers as non-compliance could result in losing license to operate in the EU market. And for players fearing stringent scrutiny in the future, operating in the European healthcare market will not be easy once the new regulations come into force.